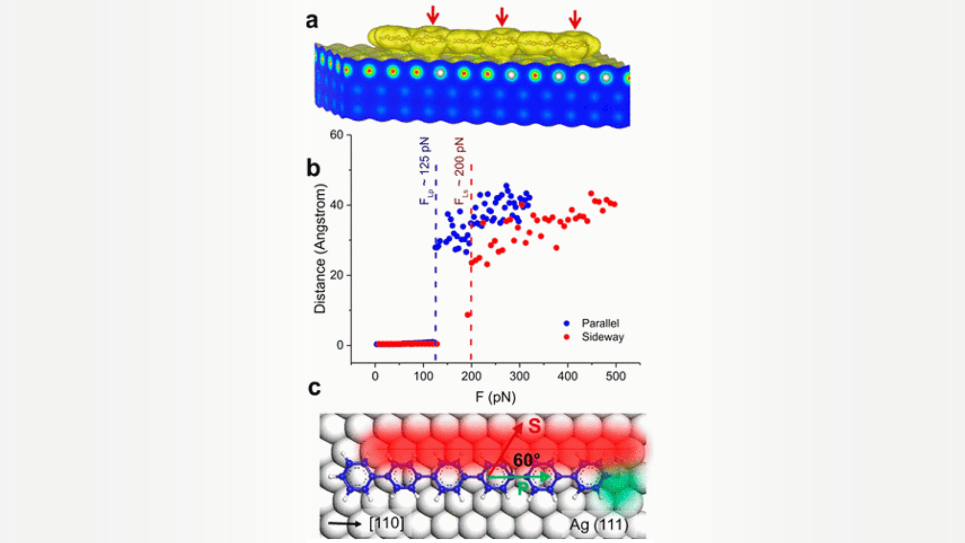
With the advancement of surface science techniques, such as atomic force microscopy (AFM) and scanning tunneling microscopy (STM), it has become possible to gain deeper insight into the lateral forces and corresponding tribological phenomena of the atoms, molecules, and nanoscale systems on surfaces. Although a myriad of studies have been conducted to elucidate the friction at the nano- and atomic scale, many tribological phenomena are yet to be understood. At the atomic scale, the potential energy landscape of surfaces plays a critical role in friction. For this experimental study, we chose para-sexiphenyl (6P) molecules because they have a representative 1D shape. 6P is composed of sixπ-rings connected as a linear chain (Figure 1b), and it is widely investigated due to potential applications in optoelectronic devices. An atomically flat Ag(111) surface, to study 6P molecules moving along the direction parallel to its molecular axis and sideways to the axis, was chosen.
To unravel the experimentally observed lateral force phenomenon, we have performed density functional theory (DFT) and classical molecular dynamics (CMD) calculations. Density Functional Theory (DFT) calculations were carried out with the Vienna ab initio simulation package (VASP) code. Geometrically relaxed DFT calculations including vdW interactions of the 6P adsorbed on a Ag(111) surface confirm the adsorption of the molecule along the [110] surface directions, in agreement with the experimental finding (Figure 1a below). The calculations also reveal twisting of alternateπ-rings (indicated with arrows in Figure 1a) with a torsional angle of∼18°.
Next, using DFT computed adsorption structure of 6P on Ag(111) as input, we performed classical molecular dynamics (CMD) simulations were performed within the canonical ensemble (NVT) to (a) identify the threshold force required to displace 6P on Ag (111) along parallel and sideways directions, as well as (b) elucidate the conformational dynamics associated with the surface sliding of 6P along these directions (under applied force). The calculations provide the threshold lateral force to displace the molecule along the parallel direction, FLP, as ∼125 pN while that along the sideways direction, FLS, as ∼200 pN. Thus, threshold lateral force along the sideways direction is larger than that of the parallel direction, and the ratio of the lateral forces, FLS/FLP, is 1.6. This ratio relatively agrees with the experiments.